Επιλέξτε χώρα ή περιοχή


-
Argentina (Español)

-
Australia (English)

-
Austria (Deutsch)

-
Bahrain (العربية)

-
Bahrain (English)
-
België (Nederlands)

-
Belgique (Français)
-
Brazil (Portugues)

-
Canada (English)

-
Canada (Français)
-
Chile (Español)

-
Colombia (Español)

-
Croatia (Croatian)

-
Denmark (Danish)

-
Deutschland (Deutsch)

-
Europe (English)

-
France (Français)

-
Greece (Ελληνικά)
-
Italia (Italiano)

-
Hungary (Magyar)

-
Lietuva (Lietuviškai)

-
Mexico (Español)

-
日本 (日本語)

-
대한민국 (한국어)

-
Kuwait (العربية)

-
Kuwait (English)
-
Nederland (Nederlands)

-
Norge (Norsk)

-
Oman (العربية)

-
Oman (English)
-
Polska (Polskie)

-
Portugal (Portuguese)

-
Qatar (العربية)

-
Qatar (English)
-
Saudi Arabia (العربية)

-
Saudi Arabia (English)
-
Slovakia (Slovak)

-
Slovenia (Slovenščina)

-
Spain (Español)

-
Suomi (Suomi)

-
Sverige (Svenska)

-
Schweiz (Deutsch)

-
台灣 (中文)

-
United States (English)

-
UAE (العربية)

-
UAE (English)
ΤΙ ΕΙΝΑΙ Η ΝΩΤΙΑΙΑ ΜΥΪΚΗ ΑΤΡΟΦΙΑ (SMA);
H SMA επηρεάζει το τμήμα του νευρικού συστήματος που ελέγχει την εκούσια μυϊκή κίνηση.1 Στη νωτιαία μυϊκή ατροφία, υπάρχει απώλεια σημαντικών κυττάρων στο νωτιαίο μυελό – που ονομάζονται κινητικοί νευρώνες – οι οποίοι είναι απαραίτητοι για τη μυϊκή δύναμη και την κίνηση. Αυτοί οι κινητικοί νευρώνες ρυθμίζουν τη μυϊκή δραστηριότητα αποστέλλοντας σήματα από το κεντρικό νευρικό σύστημα (ΚΝΣ), το οποίο είναι το μέρος του νευρικού συστήματος του σώματος που περιλαμβάνει τον εγκέφαλο και τον νωτιαίο μυελό.1,2
Η απώλεια των λειτουργικών κινητικών νευρώνων οδηγεί σε προοδευτική μυϊκή αδυναμία και ατροφία (η σταδιακή μείωση της μάζας και της δύναμης των μυών), καθώς οι μύες σταματούν να λαμβάνουν σήματα από το ΚΝΣ.3
ΚΑΤΑΝΟΗΣΗ ΤΗΣ ΑΙΤΙΟΛΟΓΙΑΣ ΤΗΣ ΝΩΤΙΑΙΑΣ ΜΥΪΚΗΣ ΑΤΡΟΦΙΑΣ
Σε αντίθεση με πολλές άλλες σπάνιες νευρομυϊκές νόσους, υπάρχει σαφής κατανόηση της ειδικής γενετικής αιτιολογίας της. Η SMA προκαλείται από μια μετάλλαξη στο γονίδιο του κινητικού νευρώνα επιβίωσης 1 (SMN1), το οποίο είναι υπεύθυνο για την παραγωγή της πρωτεΐνης του κινητικού νευρώνα επιβίωσης (SMN). Αυτή η πρωτεΐνη διατηρεί την υγεία και τη φυσιολογική λειτουργία των κινητικών νευρώνων.
Στα άτομα με νωτιαία μυϊκή ατροφία, και τα δύο αντίγραφα του γονιδίου SMN1 παρουσιάζουν μετάλλαξη, οδηγώντας σε μειωμένη παραγωγή πρωτεΐνης SMN. Χωρίς κατάλληλο επίπεδο πρωτεΐνης SMN, οι κινητικοί νευρώνες στον νωτιαίο μυελό θα χαθούν, αποτρέποντας έτσι τους μύες να λαμβάνουν σωστά σήματα από τον εγκέφαλο.4
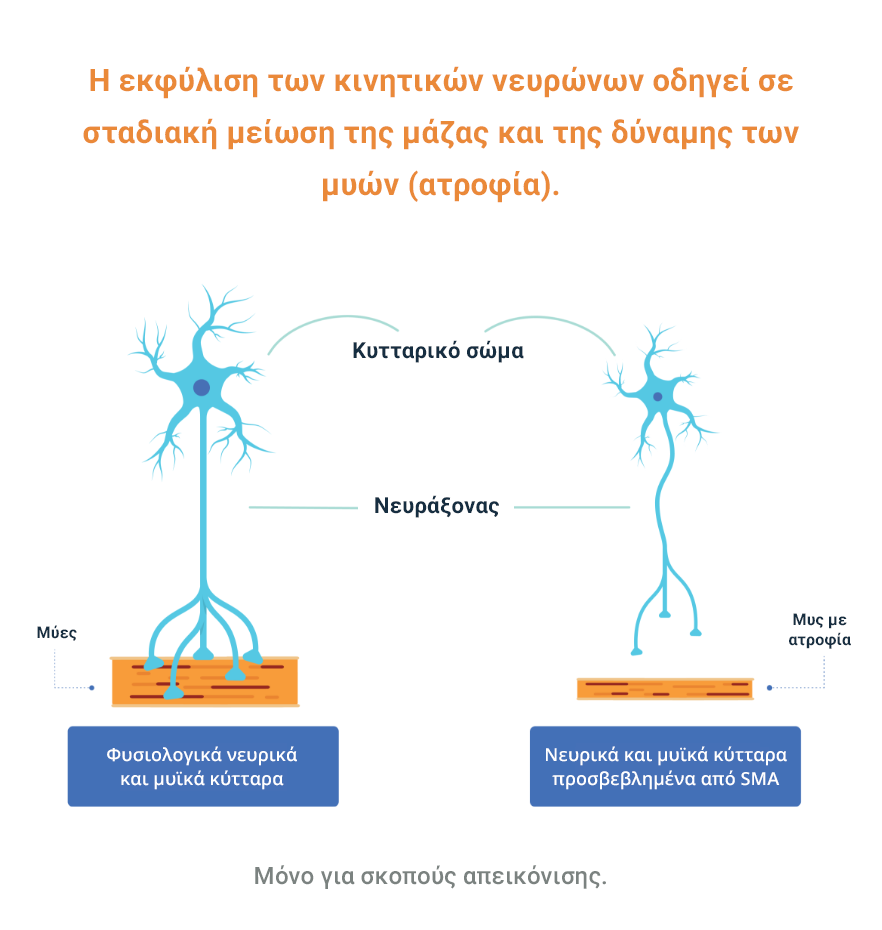
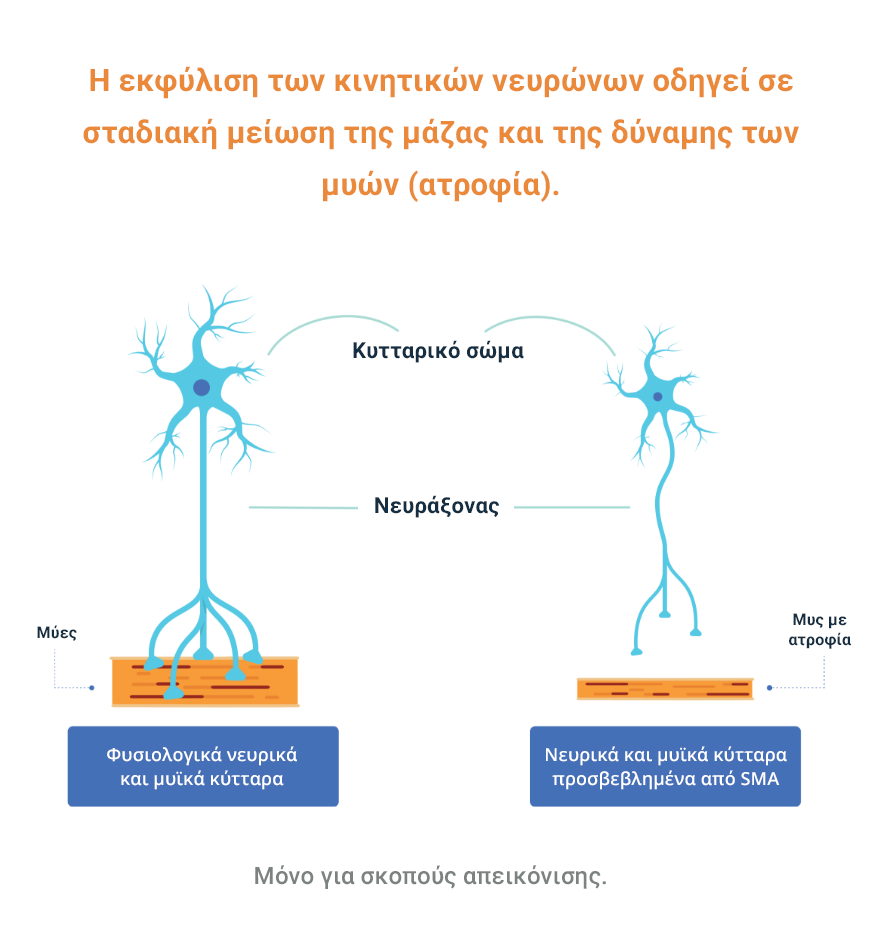
Η SMA χωρίζεται σε διαφορετικούς τύπους ανάλογα με την ηλικία έναρξης και τη λειτουργική ικανότητα. Υπάρχει επίσης ένα εύρος βαρύτητας σε κάθε τύπο.5


ΣΥΝΔΕΘΕΙΤΕ
για να μάθετε περισσότερα σχετικά με το πώς να συμμετέχετε στην κοινότητα SMA
Οι χαρακτήρες που εμφανίζονται είναι πραγματικοί ασθενείς και έχει ληφθεί η απαιτούμενη συγκατάθεση για τη χρήση των ιστοριών τους από τους ίδιους και τις οικογένειές τους. Οι φωτογραφίες προορίζονται μόνο για επεξηγηματικούς σκοπούς.
Βιβλιογραφικές αναφορές
1. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133.
2.Wang CH, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027-1049.
3. National Institute of Neurological Disorders and Stroke. Motor Neuron Disease Fact Sheet. 2012.
Available at: https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Motor-Neuron-Diseases-Fact-Sheet. Accessed January 9, 2017.
4. Genetics Home Reference. SMN2 gene. 2012. Available at: https://ghr.nlm.nih.gov/gene/SMN2. Accessed January 9, 2017.
5. Kolb SJ, Kissel JT. Spinal muscular atrophy. Arch Neurol. 2011;68(8):979-984.